

Introduction
Materials and Methods
Soil Sampling
Amplicon Sequencing Analysis
Soil Enzyme Activities
Relationship between soil microbial communities and enzyme activity
Results
Microbial community
Enzyme activity
Relationship between enzyme activity and microbial community
Predicted metabolic functions
Discussion
Introduction
Agricultural productivity and sustainability are fundamental to global food security. However, concerns about the long-term impacts of conventional farming on soil health have driven interest in alternative approaches, particularly the OMS. Soil health, a cornerstone of agricultural productivity, depends on complex interactions among soil microbiomes (e.g., mycorrhizae, bacteria, and other fungi), enzyme activity, and soil chemistry. Understanding how these contrasting managements approaches affect soil health is crucial for advancing sustainable agriculture (Reeve et al., 2016; Gamage et al., 2023).
Chestnut is a fruit tree of the genus Castanea, which grows in temperate regions of the Northern Hemisphere. There are four cultivated species: Japanese chestnut (Castanea crenata Siebold & Zucc.), Chinese chestnut (C. mollissima Blume), European chestnut (C. sativa L.), and American chestnut (C. dentata Borkh.). The Tamba region, spanning Osaka, Kyoto, and Hyogo prefectures, is known for its development of chestnut varieties and cultivation techniques, particularly after the Edo period, which subsequently spread across Japan (Nishio et al., 2011; Pereira-Lorenzo et al., 2012).
Recent studies have highlighted the impact of soil enzymes on agricultural productivity (Alkorta et al., 2003). Enzymes such as phosphatases, cellulases, and dehydrogenases mediate key biochemical processes, converting complex organic compounds into plant-available nutrients. In agricultural soils, enzyme activity is influenced by soil management practices, including the application of fertilizers, pesticides, and organic amendments (Rao et al., 2014; Neemisha and Sharma 2022). Despite these insights, the dynamics of enzyme activity under OMS and CMS, as well as their implications for soil health, are not fully understood. Organic amendments, including animal manure and green manure crops, are thought to stimulate microbial biomass and enhance enzyme activity by increasing organic matter; however, the relationship between specific microbial groups and enzyme activities remains poorly understood (Antonious et al., 2020).
Soil microorganisms are essential for biological transformations and the cycling of carbon and nitrogen in the soil, with activities that strengthen soil-plant-microorganism interactions, supported by the microbial diversity within the soil system (Hamel et al., 2006; Cruz et al., 2009). The soil microbiome, comprising symbiotic mycorrhizal fungi, nitrogen-fixing bacteria, and organic matter-decomposing fungi, forms the foundation for many essential soil functions. These microbial communities are crucial for organic matter decomposition, nutrient mineralization, and pathogen suppression, all of which are vital for plant health (Böhme et al., 2005; Bogati and Walczak, 2022).
In the case of chestnut trees, the ectomycorrhizae is ubiquitous in these roots, although AMF and bacteria have been found in these soils too. Furthermore, other studies have detected dominance of these associations over AMF, even though still reporting occasional AMF colonization in rhizosphere soils of these fruit trees (Palmer et al., 2008).
OMS enhance biotic abundance, biodiversity, soil carbon content (Gamage et al., 2023). Compared to CMS, OMS exhibit less variability in biotic abundance and richness but show greater variability in yield and require more physical effort (Gomiero et al., 2011). Even when cultivating the same crop in the same location, OMS employ a variety of nutrient management practices. This heterogeneity may help explain some of the contrasting results between landscape-scale comparisons of OMS and CMS and site-specific experiments.
Despite its advantages, OMS also presents challenges, especially concerning on the heterogeneity in management systems, variations in soil quality, and sometimes lower productivity compared to CMS. A lack of understanding persists concerning how heterogeneity affects soil microbial activity and community composition, as well as its implications for soil ecosystem functions and agroecosystem management. Microorganisms and their enzymes are crucial for maintaining soil quality, health, and carbon sequestration (Neemisha and Sharma, 2022).
This study aims to compare the effects of CMS and OMS on chestnut soil health. The microbial composition (mycorrhiza, bacteria, fungi), enzyme activity, metabolic functions, and soil nutrient levels were evaluated under different management systems to identify key differences in soil biochemical processes and microbial interactions. Additionally, wild chestnut orchards were included as a positive control to provide a natural reference for soil health. The hypothesis raised was that OMS support a more diverse and functionally active microbial community, promoting greater enzyme activity and nutrient cycling compared to CMS, thereby enhancing soil health. This hypothesis is grounded in the premise that organic amendments and practices foster beneficial microbial populations and enzymes essential for maintaining soil health.
Materials and Methods
Soil Sampling
Soil samples were collected at a depth of 10 cm beneath the canopy of chestnut trees in orchards managed under three different cultivation systems CMS, OMS and WS across four locations in the Tamba region (Wachi, Ayabe, Fukuchiyama, and Sasayama), Japan, in June 2018. The samples were stored in vacuum-sealed packs in a refrigerator at 4°C for enzyme analysis and in a freezer at -18°C for molecular analysis. Sampling was conducted with four replicates, totaling 48 samples. The soil chemical characteristics, and the management of each system, including the soil inputs, weed pest and control, and productivity were previously reported (Dilzahan et al., 2021).
Amplicon Sequencing Analysis
Soil DNA was extracted following the methodology described by (Kageyama et al., 2003), with purification carried out using the FastGeneTM Gel/PCR Extraction Kit (Nippon Genetics).
Amplicon PCR was performed using the primers 341F/785R (targeting the V3 - V4 hypervariable regions of the 16S rRNA gene) (Klindworth et al., 2013) for bacterial communities and FLR2-FLD3 primers (Niwa et al., 2018) for arbuscular mycorrhizal fungi. For bacteria, the amplicon PCR was performed with 2 μL of genomic DNA, 10 μL of KAPA HiFi HotStart Ready Mix, (Nippon Genetics Co., Japan) 4 μL of each primer (V3 and V4) (0.2 μM - final concentration in the reaction). The thermal cycler program was set as follows: 95°C for 3 min, followed by 25 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 5 min, before being held at 12°C. For arbuscular mycorrhizal fungi, 1 μL of genomic DNA, 10 μL of KOD FX Neo buffer, 0.4 μL of KOD FX Neo (Toyobo Co., Japan), 2 μL of each primer (0.5 μM - final concentration in the reaction), 4 μL of dNTPs, and 0.6 μL of water were mixed. The thermal cycler program was set as follows: 94°C for 2 min, followed by 30 cycles of 98°C for 10 s, 55°C for 30 s, and 68°C for 1 min, with a final hold at 12°C.
Index PCR and library construction were performed using S502-S522 as the forward primer and N712-N716 (Illumina, Inc.) as a reverse primer, with the method for the amplicon PCR. The middle phase was conducted with 8 and 12 cycles for bacteria and arbuscular mycorrhizal fungi, respectively. PCR products were purified using the same method for the amplicon PCR. The library was sequenced using the Illumina MiSeq 250 bp platform (Genome Quebec, Canada). Bioinformatic analysis of the sequence data was performed using QIIME2-2022 (Bolyen et al., 2018), which was used to calculate relative abundance (RA) and microbial richness (Number of Operational Taxonomic Units - OTU, and Shannon-Wiener index). The RA of bacteria was added at class level and the AMF at genus level, using the average values, the Tukey test was used to calculate the significance among the RA, although the standard errors were not inserted in the graphs. For the bacteria the Silva database (https://www.arb-silva.de) and for the AMF the MaarjAM (https://maarjam.ut.ee) (Öpik et al., 2010). The predicted metabolic functions were analyzed using bacterial data at the species level via FAPROTAX (Louca et al., 2016).
Soil Enzyme Activities
Dehydrogenase activity was measured using triphenyltetrazolium chloride to quantify triphenylformazan (Tabatabai, 1994). Phosphatase, arylsulfatase, and β-glucosidase activities were measured by converting their respective substrates into p-nitrophenol (Tabatabai, 1994). Urease activity was measured by the conversion of urea into ammonia. A 2.5 g soil sample and 1.3 mL of 0.08 N urea solution were placed in a test tube and shaken. After being sealed with rubber stoppers, the tubes were incubated at 37°C for 2 h. Then, 5 mL of HCl-KCl buffer were added and mixed thoroughly. The solution was transferred to a glass bottle, rinsing the inside of the test tube with 20 mL of HCl-KCl buffer. The solution was then shaken at 40 rpm for 30 min. After shaking, the solution was filtered, and the filtrate was analyzed for absorbance using the indophenol method with 2-hydroxybiphenyl sodium salt (Tabatabai, 1994).
Protease activity was measured by adding benzyloxycarbonyl-phenylalanine-leucine (ZFL) to soil samples (Yamagata et al., 1997). Amilase, invertase and cellulase activities were evaluated by adding their respective substrates to soil samples and measuring glucose formation (Schinner and von Mersi, 1990). Geometric Mean of Enzyme Activity (GEA) was calculated using the formula:
where M, N, O, P, Q, R, S, T, and U represent the enzymes analyzed with their respective units (García-Ruiz et al., 2008; Mierzwa-Hersztek et al., 2019).
Relationship between soil microbial communities and enzyme activity
The general correlation between all data from the soil microbial communities (relative abundance) and enzyme activities, was analyzed using Non-Metric Multidimensional Scaling (NMDS). Such analysis was conducted using enzyme activities and the number of OTUs within the soil microbial communities as variables, employing the ggplot2, ggrepel, and vegan packages in the statistical software RStudio 1.4. The fungal community data from the same plots (Dilzahan et al., 2021) were used to calculate NMDS, complementing the information of this study. The Tukey test was used for statistical analysis to calculate the significance between treatments.
Results
Microbial community
Results on bacterial relative abundance showed that the most common bacterial classes were Alphaproteobacteria, Betaproteobacteria, and Gammaproteobacteria. Compared to other samples, soils under OMS at Sasayama exhibited a higher abundance of Nitrospira. A greater value of the class Bacilli was detected under WS in Fukuchiyama and Ayabe, whereas the Flavobacteriia were typically found under OMS at Wachi and Ayabe (Fig. 1A).
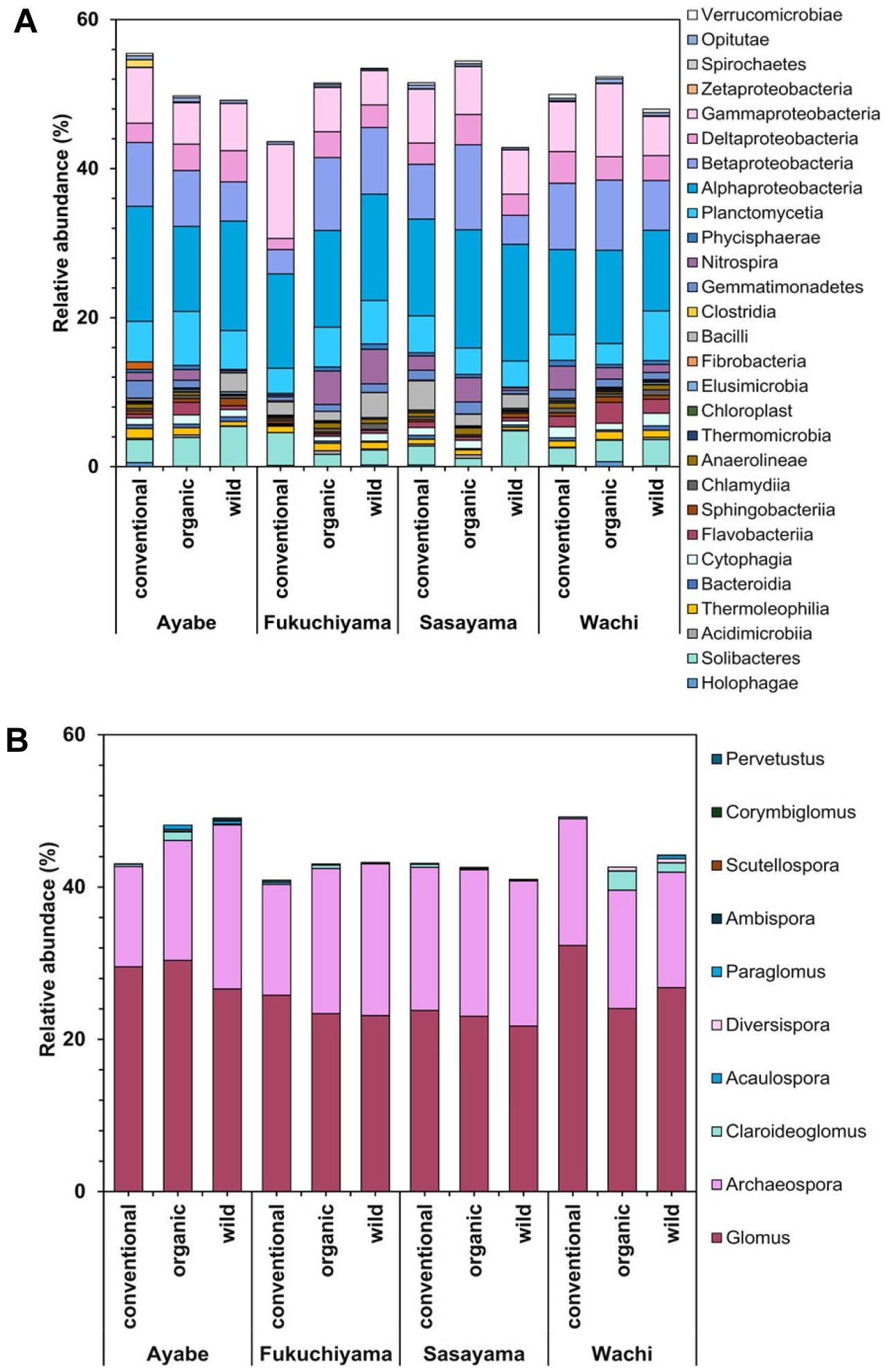
The relative abundance of AMF indicated that the genus Glomus and Archaeospora were dominant in all orchards. The Claroideoglomus was detected in Ayabe and Wachi, while the Paraglomus was observed in Ayabe under OMS. Other genus were relatively specific to certain management types, such as the Claroideoglomus, which was specific to orchards at Wachi and Ayabe under OMS (Fig. 1B).
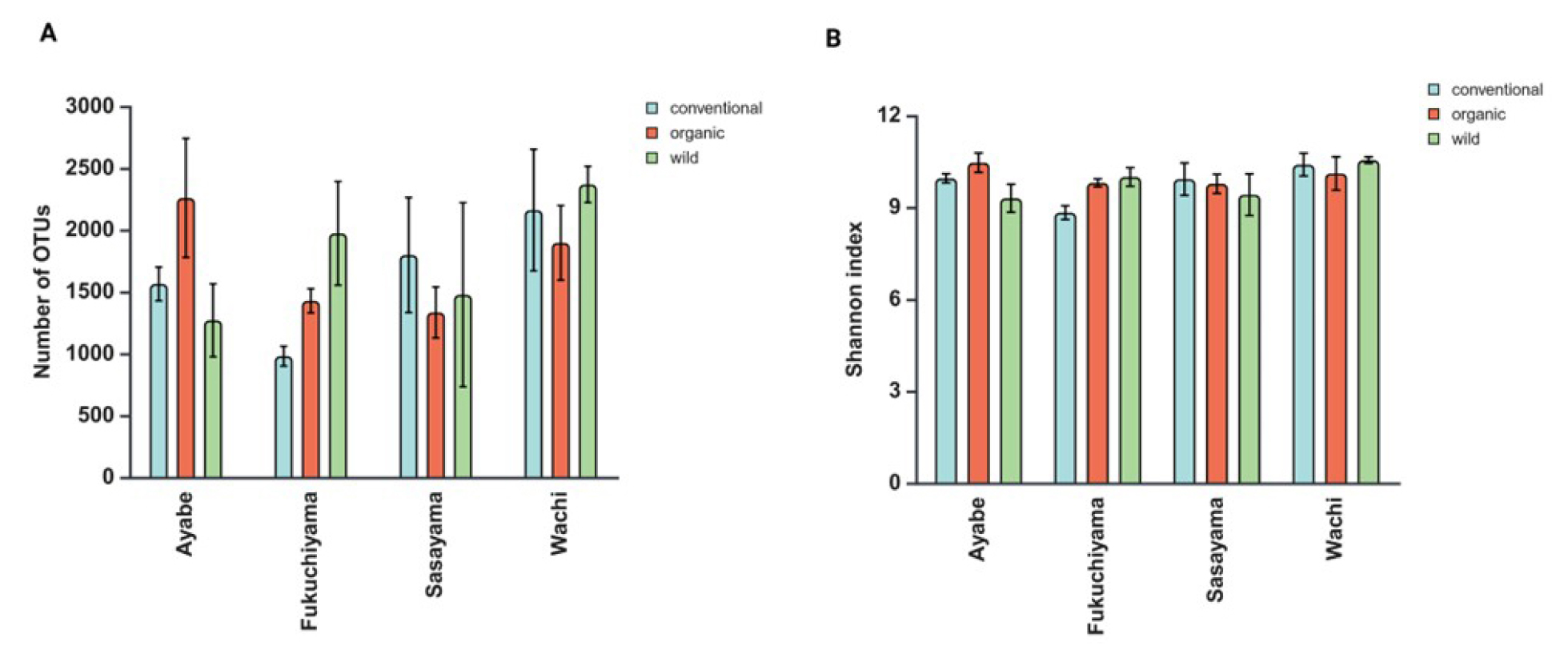
The number of OTUs was higher under OMS bat Ayabe and Fukuchiyama (Fig. 2A). The SW was higher under OMS and WS at Fukuchiyama and Ayabe (Fig. 2B). The AMF richness that was evaluated based on number of OTUs and the SW, did not differ among systems and locations (data not shown).
Enzyme activity
Enzyme activities indicated that the GEA formed a gradient growth in this sequence (CMS-OS-WS), although no statistical differences observed among them (Fig. 3A). Furthermore, for most of them the WS exhibited the highest enzyme activity, followed by OMS and CMS. However, arylsulfatase, β-glucosidase, and invertase activities were higher under OMS as compared to CMS (Fig. 3B).
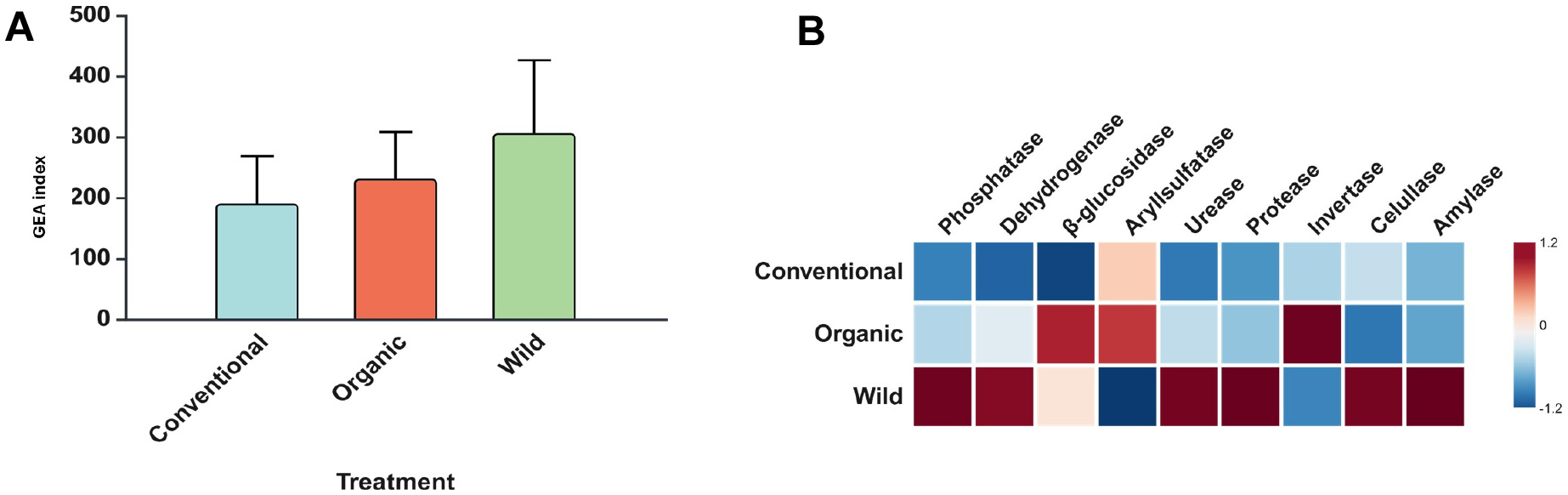
Fig. 3
Geometric Mean of Enzyme Activity (A) and heatmap containing the activity of nine enzymes (B) in sampled areas separated by three cultivation systems. The values were normalized using z-scores, calculated as (x - μ)/σ, where x is the raw value, μ is the mean, and σ is the standard deviation. Rows represent management systems, and columns represent enzymes. The color scale indicates the relative intensity of the z-scores, with blue representing negative z-scores (below the mean), white representing values near the mean, and red representing positive z-scores (above the mean).
Relationship between enzyme activity and microbial community
The NMDS data showed that the activities of some enzyme were generally influenced by the microbial community. Dehydrogenase, phosphatase, cellulase, urease, protease, and GEA were significantly affected in bacteria (Fig. 4A). A similar result was found for fungi and AMF, which also included amylase (Fig. 4B, C). WS exhibited the strongest effect in driving these relationships, followed by OMS and CMS (Fig. 4).
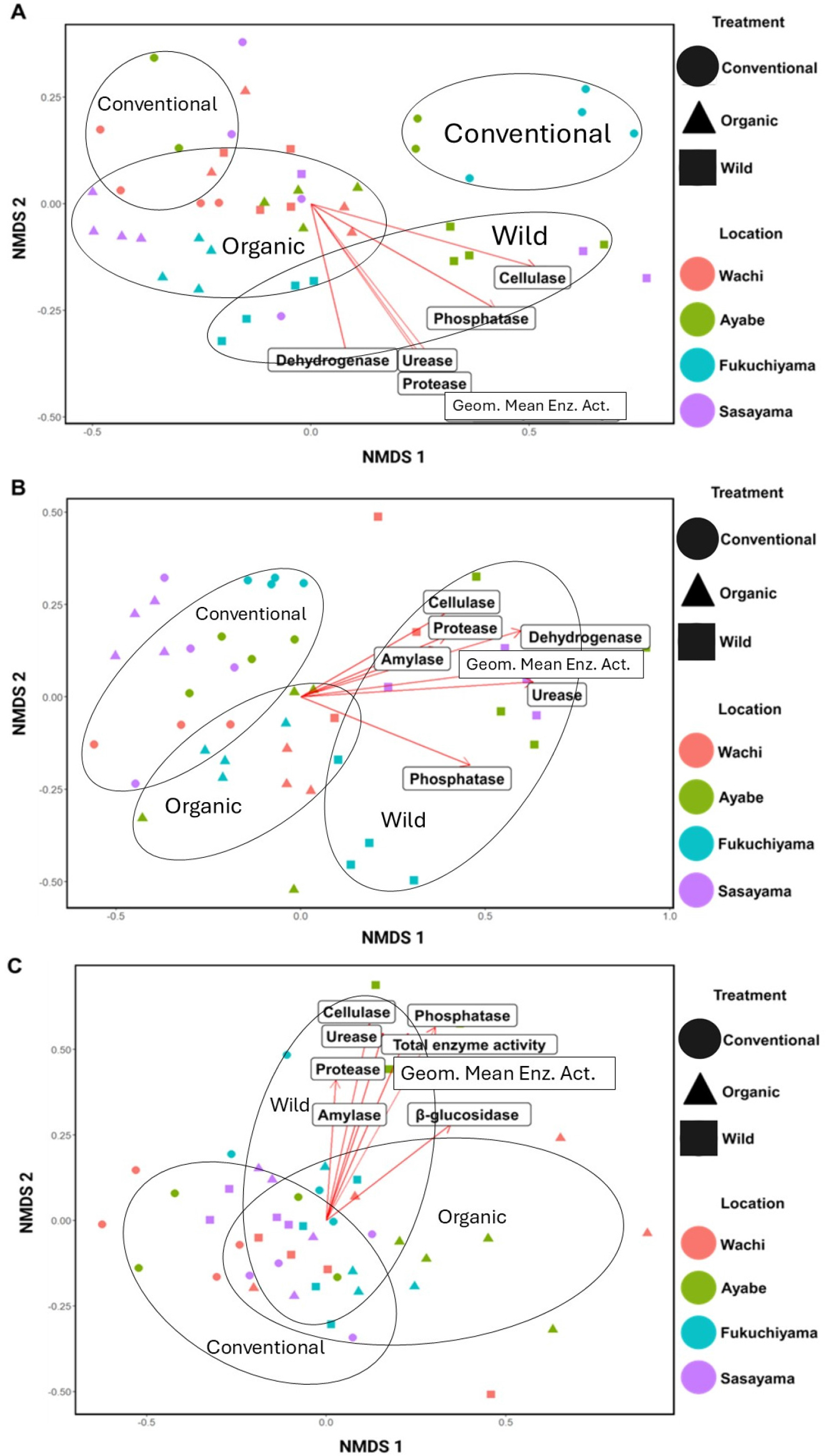
Fig. 4
Non-Metric Multidimensional Scaling (NMDS) ordination of microbial and enzymatic activity relationships. The NMDS plots illustrate the relationships between microbial community composition and enzymatic, activities for (A) Bacterial communities, (B) Fungal communities, and (C) Mycorrhizal communities. The ordination was based on Bray-Curtis dissimilarity for community composition enzyme activity vectors fitted to the ordination. Points represent samples and colors or shapes indicate treatments. Enzyme vectors are shown as arrows, with their direction and length indicating the strength and correlation of enzymatic activities with the ordination axes.
Predicted metabolic functions
Regarding functions related to N cycling, OMS exhibited the highest values for most of them, except for the nitrate reduction and ureolysis. WS exhibited the lowest levels of ammonia oxidation, denitrification and N respiration. These functions had the lowest values under CMS, except for ureolysis (Fig. 5). For functions related to C cycling, CMS had the highest values for plant pathogens, photoautotrophy, photoheterotrophy, and fermentation, whereas WS was characterized by xylanolysis and chemoheterotrophy. Cellulolysis was higher under OMS (Fig. 5).
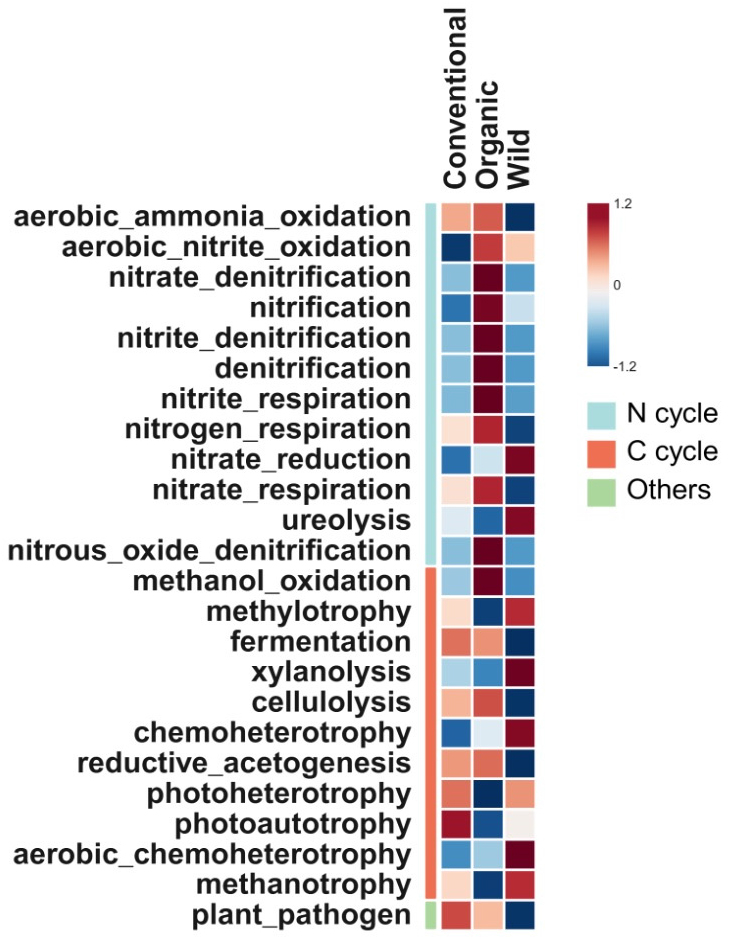
Fig. 5
Predicted metabolic functions (FAPROTAX) from bacterial community among the cultivation systems. The values were normalized using z-scores, calculated as (x - μ)/σ, where x is the raw value, μ is the mean, and σ is the standard deviation. Rows represent management systems, and columns represent enzymes. The color scale indicates the relative intensity of the z-scores, with blue representing negative z-scores (below the mean), white representing values near the mean, and red representing positive z-scores (above the mean).
Discussion
This study emphasizes the impact of agricultural practices on soil microbial communities and enzyme activities. OMS promoted greater microbial diversity and some enzyme activities. The organic practices can improve nitrogen cycling, soil fertility, and sustainability by fostering a diverse microbial community (Antonious et al., 2020).
The results revealed that Alphaproteobacteria and Betaproteobacteria dominated bacterial communities across various agricultural systems, with minimal variation between CMS and OMS. Additionally, the Nitrospira had a higher relative abundance under OMS. This suggests that factors beyond agricultural practices, such as soil chemistry, are crucial in shaping bacterial communities. Consistent with earlier research, bacterial community structure was influenced by a complex interplay of factors rather than solely by the direct effects of soil acidity (Lammel et al., 2018; Philippot et al., 2024).
Furthermore, soil enzyme activities, particularly β-glucosidase, invertase, and arylsulfatase, were more strongly associated with OMS, however the relationship with microbial communities showed a high influence of WS. This could indicate a stronger effects of sustainable systems in these as compared to CMS. Enzyme activity, as an indicator of soil quality, reflects the interactions between microbial communities and environmental factors (Alkorta et al., 2003; Zhang et al., 2015). OMS generally promote greater microbial diversity and enzymatic activity compared to CMS, which sometimes are associated to lower microbial diversity, that may induce the likelihood of pathogenic bacteria reaching harmful levels (Ambrosini et al., 2016; van Bruggen et al., 2016).
Glomus and Archaeospora were the most frequently detected AMF genera across the systems, and specific orchards managed under OMS exhibited distinct AMF communities. The AMF has a crucial role in the nutrient cycling in the soil, because they contribute to P solubilization, biocontrol of soil-borne plant pathogens and sometimes heavy metal contamination (Han et al., 2025). Therefore, the detection of these AMF groups in these systems may serve as indicators of organic farming practices (Purin et al., 2006; Kim et al., 2022).
The promotion of the bacteria, fungi and AMF capable of driving key nutrient cycle processes could be enhanced by environmental friendly systems, as shown by the NMDS data. The decomposition of organic amendments releases labile carbon sources that promote microbial activity, stimulating essential enzymatic processes for nutrient transformations. In such case, all microbial groups described here, could directly or non-directly contribute to the nutrient cycling, sometime the AMF hyphae function as attractive for beneficial bacteria in the soils (Cruz and Ishii, 2012). These authors affirmed that under natural conditions, the AMF hyphae sometimes attract beneficial bacteria, which could contribute to the nutrient cycling in soil. OMS promote functionally diverse microbial communities, including nitrifying bacteria such as Nitrospira which enhance N cycling efficiency (Ouyang et al., 2022). The increased activity of β-glucosidase, urease, and phosphatase under OMS indicates a microbial-driven acceleration of carbon, nitrogen, and phosphorus cycling, supporting long-term soil fertility. In the case specific of the sampled areas, the higher available P and amount of humus content (Dilzahan et al., 2021) could contribute for the C cycling and higher microbial richness, here represented by Shannon diversity and number of OTUs.
These findings are consistent with studies demonstrating that organic management practices enhance soil microbial diversity and function, leading to improved nutrient cycling and soil health, and highlight that OMS promote nutrient cycling, which benefits soil fertility and agricultural sustainability (Mohan et al., 2021; Yu et al., 2024). The observed increase in microbial activity is consistent with these studies, which suggest that organic amendments significantly influence microbial functions related to nutrient cycling. Moreover, nitrogen management is crucial for enhancing soil health (Rodríguez-Espinosa et al., 2023).
Both OMS and WS could contribute to more sustainable and resilient soil management practices by promoting microbial diversity, improving nitrogen and carbon cycling (Brown et al., 2022). Further investigation across various agricultural systems is needed to refine management practices and optimize soil health and sustainability.
This study highlights the importance of agricultural practices in shaping soil microbial communities and enzyme activities. Organic management systems enhance microbial diversity and enzyme activity, while contributing to improved nitrogen and carbon cycling. Wild followed by organic soils have distinct nitrogen retention mechanisms, higher microbial diversity, and drive the relationship between enzyme activity and the microbial community, represented by the relative abundance. This can emphasizing the value of undisturbed ecosystems in maintaining balanced nutrient cycles. Overall, the findings suggest that organic management systems and wild systems promote more resilient and sustainable soil health compared to conventional management systems, with regional soil variations significantly influencing microbial and nutrient dynamics.
